
Chromatogram File Utils
For Sanger sequencing data visualizing, alignment, mutation calling, and trimming etc.
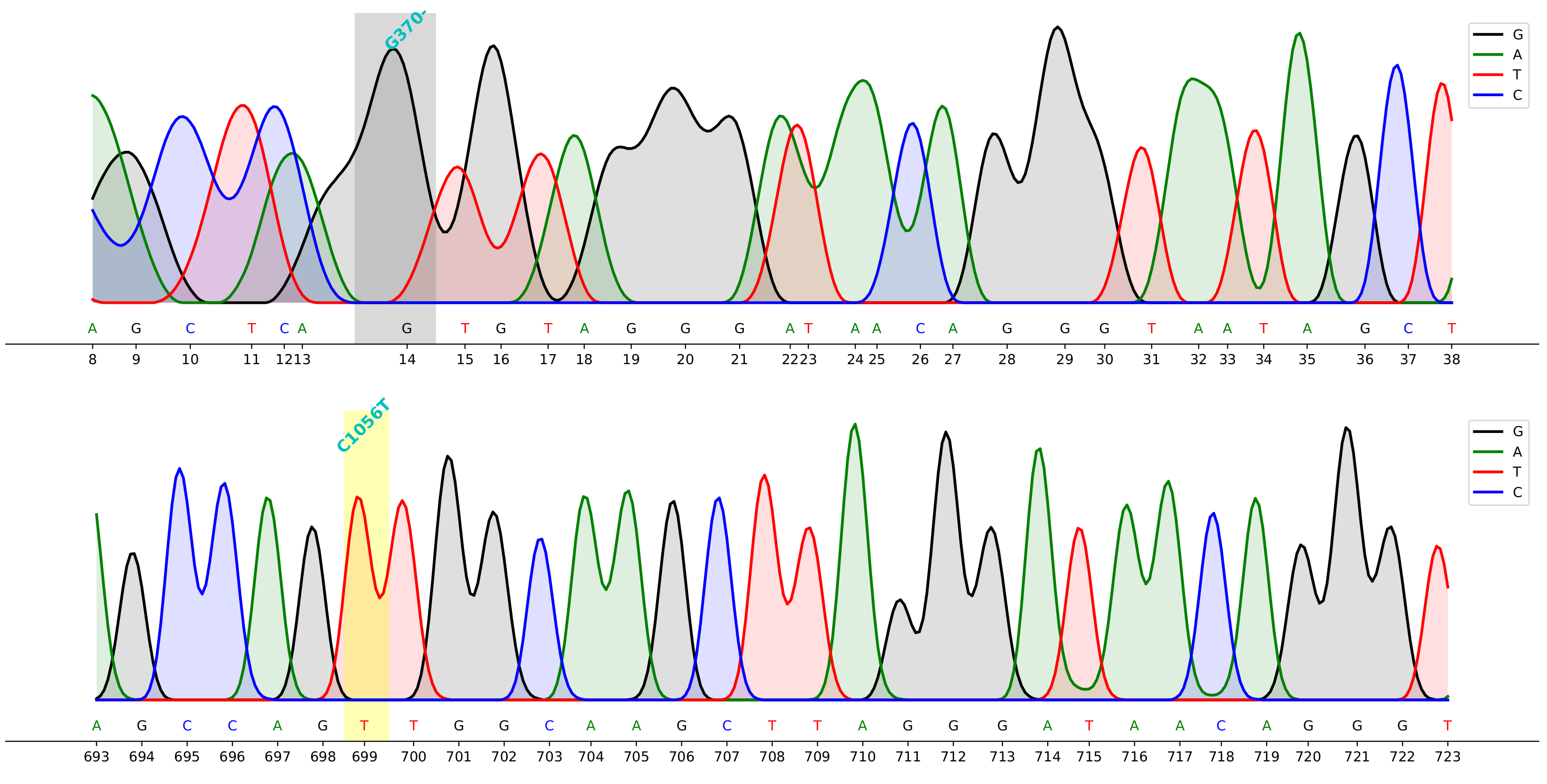
command to generate the demo above
cfutils mut --query ./data/B5-M13R_B07.ab1 --subject ./data/ref.fa --outdir ./data/ --plot- You can have mutation detection and visualization in one step using the command line.
cfutils mut --help- You can also integrate the result matplotlib figures and use it as a python module.
An example:
import matplotlib.pyplot as plt
import numpy as np
from cfutils.parser import parse_abi
from cfutils.show import plot_chromatograph
seq = parse_abi("./data/B5-M13R_B07.ab1")
peaks = seq.annotations["peak positions"][100:131]
fig, axes = plt.subplots(2, 1, figsize=(12, 6), sharex=True)
plot_chromatograph(
seq,
region=(100, 130),
ax=axes[0],
show_bases=True,
show_positions=True,
color_map=dict(zip("ATGC", ["C0", "C2", "C1", "C4"])),
)
axes[1].bar(peaks, np.random.randn(len(peaks)), color="0.66")
plt.show()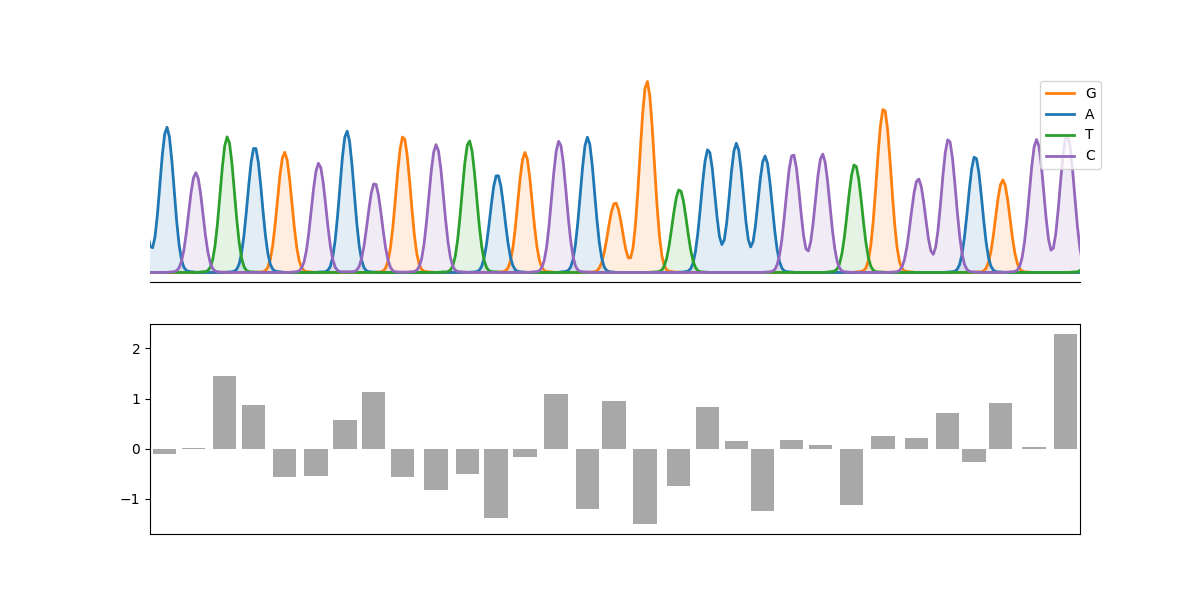
(use this way ONLY, if you don't know what's going on)
pip install --user cfutils- clone from github
git clone [email protected]:y9c/cfutils.git- install the dependence
make init- do unittest
make test- Reverse completement the chromatogram file. (Inspired by Snapgene)
- build as python package for pypi
- fix bug that highlighting wrong base
- replace blastn with buildin python aligner
-
call mutation by alignment and plot Chromatogram graphic
-
add a doc
-
change xaxis by peak location
-
fix bug that chromatogram switch pos after trim
-
wrap as a cli app
-
return quality score in output
-
fix issue that selected base is not in the middle
-
fix plot_chromatograph rendering bug
-
add projection feature to make align and assemble possible