Home
The purpose of treeplyr is to provide functions for matching a tree to data, and to manipulate that data using dplyr--all while maintaining a perfect match between the tree and the data.
First we'll load some packages and generate some data to analyze. We're going to create a somewhat messy dataset and use treeplyr to manipulate the data to fit our needs.
require(treeplyr)
tree <- geiger::sim.bdtree(stop="taxa", n=50, seed=1)
dat <- data.frame(X1 = rnorm(50), X2 = rnorm(50), X3 = rnorm(50),
taxa=c(tree$tip.label[1:40], paste("s", 51:60, sep="")),
D1=sample(c("Hello", "World"), 50, replace=TRUE), D2=rbinom(50,1,0.5),
D3=0.3+rbinom(50, 4, c(0.1,0.1,0.5,0.3)), XNA1 = c(rep(NA, 10), rnorm(40)))Let's check and see what the tree and data look like:
tree##
## Phylogenetic tree with 50 tips and 49 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
head(dat)## X1 X2 X3 taxa D1 D2 D3 XNA1
## 1 -0.5425200 -1.9143594 -0.1771040 s1 World 1 0.3 NA
## 2 1.2078678 1.1765833 0.4020118 s2 Hello 0 2.3 NA
## 3 1.1604026 -1.6649724 -0.7317482 s3 World 0 2.3 NA
## 4 0.7002136 -0.4635304 0.8303732 s4 World 0 0.3 NA
## 5 1.5868335 -1.1159201 -1.2080828 s5 World 0 0.3 NA
## 6 0.5584864 -0.7508190 -1.0479844 s6 World 0 0.3 NA
Notice that the dataset we have created has a mix of trait types, with both discrete and continuous characters, some traits with missing data, and species names buried in the middle of the data matrix. The function make.treedata will search the data table for the column with the most matches to the tree, and automatically use this column for matching. It will also search the rownames. Either way, the command is quite simple:
td <- make.treedata(tree, dat)We can use summary to display information about our treedata object.
summary(td)## A treeplyr treedata object
## The dataset contains 7 traits
## Continuous traits: X1 X2 X3 D3 XNA1
## Discrete traits: D1 D2
## The following traits have missing values: XNA1
## These taxa were dropped from the tree: s51, s52, s53, s54, s55, s56, s57, s58, s59, s60
## These taxa were dropped from the data: s41, s42, s43, s44, s45, s46, s47, s48, s49, s50
## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 7]
##
## X1 X2 X3 D1 D2 D3 XNA1
## (dbl) (dbl) (dbl) (fctr) (int) (dbl) (dbl)
## 1 -0.5425200 -1.91435943 -0.1771040 World 1 0.3 NA
## 2 1.2078678 1.17658331 0.4020118 Hello 0 2.3 NA
## 3 1.1604026 -1.66497244 -0.7317482 World 0 2.3 NA
## 4 0.7002136 -0.46353040 0.8303732 World 0 0.3 NA
## 5 1.5868335 -1.11592011 -1.2080828 World 0 0.3 NA
## 6 0.5584864 -0.75081900 -1.0479844 World 0 0.3 NA
## 7 -1.2765922 2.08716655 1.4411577 Hello 0 1.3 NA
## 8 -0.5732654 0.01739562 -1.0158475 Hello 0 1.3 NA
## 9 -1.2246126 -1.28630053 0.4119747 World 0 2.3 NA
## 10 -0.4734006 -1.64060553 -0.3810761 Hello 1 1.3 NA
## .. ... ... ... ... ... ... ...
We can also use indices directly on the treedata object, but note that these drop the tree:
td[[1]]## s1 s2 s3 s4 s5 s6
## -0.54252003 1.20786781 1.16040262 0.70021365 1.58683345 0.55848643
## s7 s8 s9 s10 s11 s12
## -1.27659221 -0.57326541 -1.22461261 -0.47340064 -0.62036668 0.04211587
## s13 s14 s15 s16 s17 s18
## -0.91092165 0.15802877 -0.65458464 1.76728727 0.71670748 0.91017423
## s19 s20 s21 s22 s23 s24
## 0.38418536 1.68217608 -0.63573645 -0.46164473 1.43228224 -0.65069635
## s25 s26 s27 s28 s29 s30
## -0.20738074 -0.39280793 -0.31999287 -0.27911330 0.49418833 -0.17733048
## s31 s32 s33 s34 s35 s36
## -0.50595746 1.34303883 -0.21457941 -0.17955653 -0.10019074 0.71266631
## s37 s38 s39 s40
## -0.07356440 -0.03763417 -0.68166048 -0.32427027
td[['X1']]## s1 s2 s3 s4 s5 s6
## -0.54252003 1.20786781 1.16040262 0.70021365 1.58683345 0.55848643
## s7 s8 s9 s10 s11 s12
## -1.27659221 -0.57326541 -1.22461261 -0.47340064 -0.62036668 0.04211587
## s13 s14 s15 s16 s17 s18
## -0.91092165 0.15802877 -0.65458464 1.76728727 0.71670748 0.91017423
## s19 s20 s21 s22 s23 s24
## 0.38418536 1.68217608 -0.63573645 -0.46164473 1.43228224 -0.65069635
## s25 s26 s27 s28 s29 s30
## -0.20738074 -0.39280793 -0.31999287 -0.27911330 0.49418833 -0.17733048
## s31 s32 s33 s34 s35 s36
## -0.50595746 1.34303883 -0.21457941 -0.17955653 -0.10019074 0.71266631
## s37 s38 s39 s40
## -0.07356440 -0.03763417 -0.68166048 -0.32427027
td[1:10,1:2]## Source: local data frame [10 x 2]
##
## X1 X2
## <dbl> <dbl>
## 1 -0.5425200 -1.91435943
## 2 1.2078678 1.17658331
## 3 1.1604026 -1.66497244
## 4 0.7002136 -0.46353040
## 5 1.5868335 -1.11592011
## 6 0.5584864 -0.75081900
## 7 -1.2765922 2.08716655
## 8 -0.5732654 0.01739562
## 9 -1.2246126 -1.28630053
## 10 -0.4734006 -1.64060553
For single brackets ('[]'), we can specify that we want to keep the tip labels:
td[1:10, 1, tip.label=TRUE]## Source: local data frame [10 x 2]
##
## tip.label X1
## <fctr> <dbl>
## 1 s1 -0.5425200
## 2 s2 1.2078678
## 3 s3 1.1604026
## 4 s4 0.7002136
## 5 s5 1.5868335
## 6 s6 0.5584864
## 7 s7 -1.2765922
## 8 s8 -0.5732654
## 9 s9 -1.2246126
## 10 s10 -0.4734006
The treedata object itself is made up of a list of two elements $phy giving the tree and $dat providing in the data. One operation that is relatively common is changing the ordering of the phylogeny. It's important to maintain a match between the tree and the data.
td <- reorder(td, "postorder")We can use dplyr functions select, filter and mutate directly on the treedata object. The select function allows you to choose which columns you want in the dataset. Any number of columns can be specified:
select(td, X1, D1)## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 2]
##
## X1 D1
## <dbl> <fctr>
## 1 -0.5425200 World
## 2 1.2078678 Hello
## 3 1.1604026 World
## 4 0.7002136 World
## 5 1.5868335 World
## 6 0.5584864 World
## 7 -1.2765922 Hello
## 8 -0.5732654 Hello
## 9 -1.2246126 World
## 10 -0.4734006 Hello
## .. ... ...
select(td, 1:3)## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 3]
##
## X1 X2 X3
## <dbl> <dbl> <dbl>
## 1 -0.5425200 -1.91435943 -0.1771040
## 2 1.2078678 1.17658331 0.4020118
## 3 1.1604026 -1.66497244 -0.7317482
## 4 0.7002136 -0.46353040 0.8303732
## 5 1.5868335 -1.11592011 -1.2080828
## 6 0.5584864 -0.75081900 -1.0479844
## 7 -1.2765922 2.08716655 1.4411577
## 8 -0.5732654 0.01739562 -1.0158475
## 9 -1.2246126 -1.28630053 0.4119747
## 10 -0.4734006 -1.64060553 -0.3810761
## .. ... ... ...
select(td, 1, 4, 6)## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 3]
##
## X1 D1 D3
## <dbl> <fctr> <dbl>
## 1 -0.5425200 World 0.3
## 2 1.2078678 Hello 2.3
## 3 1.1604026 World 2.3
## 4 0.7002136 World 0.3
## 5 1.5868335 World 0.3
## 6 0.5584864 World 0.3
## 7 -1.2765922 Hello 1.3
## 8 -0.5732654 Hello 1.3
## 9 -1.2246126 World 2.3
## 10 -0.4734006 Hello 1.3
## .. ... ... ...
We can also use the filter function allows us to select only those rows that meet a specific critierion. Multiple criteria can be used to limit the dataset even more. note that as the dataset is filtered, the tree is automatically pruned to reflect the datasets represented.
filter(td, X1 > 0, D1=="Hello", is.na(XNA1)==FALSE)## $phy
##
## Phylogenetic tree with 6 tips and 5 internal nodes.
##
## Tip labels:
## [1] "s14" "s16" "s17" "s20" "s32" "s36"
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [6 x 7]
##
## X1 X2 X3 D1 D2 D3 XNA1
## <dbl> <dbl> <dbl> <fctr> <int> <dbl> <dbl>
## 1 0.1580288 -0.92936215 -0.33090780 Hello 0 0.3 -1.2941400
## 2 1.7672873 -1.07519230 2.49766159 Hello 0 1.3 1.3079015
## 3 0.7167075 1.00002880 0.66706617 Hello 1 0.3 1.4970410
## 4 1.6821761 1.86929062 0.51010842 Hello 1 1.3 0.4820295
## 5 1.3430388 0.10580237 0.00213186 Hello 0 1.3 0.9465856
## 6 0.7126663 -0.03472603 1.80314191 Hello 1 1.3 0.7395892
filter(td, X1 + X2 > 0 & D1 == "Hello")## $phy
##
## Phylogenetic tree with 9 tips and 8 internal nodes.
##
## Tip labels:
## s2, s7, s16, s17, s20, s32, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [9 x 7]
##
## X1 X2 X3 D1 D2 D3 XNA1
## <dbl> <dbl> <dbl> <fctr> <int> <dbl> <dbl>
## 1 1.2078678 1.17658331 0.40201178 Hello 0 2.3 NA
## 2 -1.2765922 2.08716655 1.44115771 Hello 0 1.3 NA
## 3 1.7672873 -1.07519230 2.49766159 Hello 0 1.3 1.307901520
## 4 0.7167075 1.00002880 0.66706617 Hello 1 0.3 1.497041009
## 5 1.6821761 1.86929062 0.51010842 Hello 1 1.3 0.482029504
## 6 1.3430388 0.10580237 0.00213186 Hello 0 1.3 0.946585640
## 7 -0.2145794 0.45699881 -0.63030033 Hello 0 0.3 0.004398704
## 8 0.7126663 -0.03472603 1.80314191 Hello 1 1.3 0.739589226
## 9 -0.6816605 1.02739244 0.19719344 Hello 0 2.3 -0.289499367
The function mutate adds a new variable to the dataset. It may be a transformation of one or more existing variables. For example, we may wish to log transform a variable, or average two or more variables.
mutate(td, Xall = (X1+X2+X3)/3, D1.binary = as.numeric(D1)-1)## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 9]
##
## X1 X2 X3 D1 D2 D3 XNA1 Xall
## <dbl> <dbl> <dbl> <fctr> <int> <dbl> <dbl> <dbl>
## 1 -0.5425200 -1.91435943 -0.1771040 World 1 0.3 NA -0.8779945
## 2 1.2078678 1.17658331 0.4020118 Hello 0 2.3 NA 0.9288210
## 3 1.1604026 -1.66497244 -0.7317482 World 0 2.3 NA -0.4121060
## 4 0.7002136 -0.46353040 0.8303732 World 0 0.3 NA 0.3556855
## 5 1.5868335 -1.11592011 -1.2080828 World 0 0.3 NA -0.2457231
## 6 0.5584864 -0.75081900 -1.0479844 World 0 0.3 NA -0.4134390
## 7 -1.2765922 2.08716655 1.4411577 Hello 0 1.3 NA 0.7505773
## 8 -0.5732654 0.01739562 -1.0158475 Hello 0 1.3 NA -0.5239058
## 9 -1.2246126 -1.28630053 0.4119747 World 0 2.3 NA -0.6996461
## 10 -0.4734006 -1.64060553 -0.3810761 Hello 1 1.3 NA -0.8316941
## .. ... ... ... ... ... ... ... ...
## Variables not shown: D1.binary <dbl>.
Note that if you want to use treeplyr programatically, you may not want to use these functions, as they will not result in the desired behavior. For example:
mytraits <- c('X1', 'D1')
try(select(td, mytraits))The treeplyr function expects a column called "mytraits" rather than evaluating the variable mytraits. To use programatically (as with dplyr), use the same functions followed by an underscore:
select_(td, .dots=as.list(mytraits))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 2]
##
## X1 D1
## <dbl> <fctr>
## 1 -0.5425200 World
## 2 1.2078678 Hello
## 3 1.1604026 World
## 4 0.7002136 World
## 5 1.5868335 World
## 6 0.5584864 World
## 7 -1.2765922 Hello
## 8 -0.5732654 Hello
## 9 -1.2246126 World
## 10 -0.4734006 Hello
## .. ... ...
A similar operation can be done for functions such as filter and mutate:
criteria <- list("X1 > -1", "D1 == 'Hello'", "is.na(XNA1)==TRUE")
filter_(td, .dots=criteria)## $phy
##
## Phylogenetic tree with 3 tips and 2 internal nodes.
##
## Tip labels:
## [1] "s2" "s8" "s10"
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [3 x 7]
##
## X1 X2 X3 D1 D2 D3 XNA1
## <dbl> <dbl> <dbl> <fctr> <int> <dbl> <dbl>
## 1 1.2078678 1.17658331 0.4020118 Hello 0 2.3 NA
## 2 -0.5732654 0.01739562 -1.0158475 Hello 0 1.3 NA
## 3 -0.4734006 -1.64060553 -0.3810761 Hello 1 1.3 NA
treeplyr is mostly just a wrapper that passes on functions to dplyr, so most of the dplyr functionality is still there. All treeplyr does is make sure the tree and data stay matched through the course of your data manipulations. In other words, you can still combine select, mutate, and filter with lots of other nice dplyr functions. Here are some examples:
select(td, starts_with("D"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 3]
##
## D1 D2 D3
## <fctr> <int> <dbl>
## 1 World 1 0.3
## 2 Hello 0 2.3
## 3 World 0 2.3
## 4 World 0 0.3
## 5 World 0 0.3
## 6 World 0 0.3
## 7 Hello 0 1.3
## 8 Hello 0 1.3
## 9 World 0 2.3
## 10 Hello 1 1.3
## .. ... ... ...
select(td, ends_with("1"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 3]
##
## X1 D1 XNA1
## <dbl> <fctr> <dbl>
## 1 -0.5425200 World NA
## 2 1.2078678 Hello NA
## 3 1.1604026 World NA
## 4 0.7002136 World NA
## 5 1.5868335 World NA
## 6 0.5584864 World NA
## 7 -1.2765922 Hello NA
## 8 -0.5732654 Hello NA
## 9 -1.2246126 World NA
## 10 -0.4734006 Hello NA
## .. ... ... ...
select(td, matches("NA"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## # A tibble: 40 x 1
## XNA1
## <dbl>
## 1 NA
## 2 NA
## 3 NA
## 4 NA
## 5 NA
## 6 NA
## 7 NA
## 8 NA
## 9 NA
## 10 NA
## # ... with 30 more rows
select(td, contains("NA"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## # A tibble: 40 x 1
## XNA1
## <dbl>
## 1 NA
## 2 NA
## 3 NA
## 4 NA
## 5 NA
## 6 NA
## 7 NA
## 8 NA
## 9 NA
## 10 NA
## # ... with 30 more rows
select(td, which(sapply(td$dat, type_sum)=="int"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## # A tibble: 40 x 1
## D2
## <int>
## 1 1
## 2 0
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 1
## # ... with 30 more rows
Or dropping columns:
select(td, -matches("NA"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## # A tibble: 40 x 6
## X1 X2 X3 D1 D2 D3
## <dbl> <dbl> <dbl> <fctr> <int> <dbl>
## 1 -0.5425200 -1.91435943 -0.1771040 World 1 0.3
## 2 1.2078678 1.17658331 0.4020118 Hello 0 2.3
## 3 1.1604026 -1.66497244 -0.7317482 World 0 2.3
## 4 0.7002136 -0.46353040 0.8303732 World 0 0.3
## 5 1.5868335 -1.11592011 -1.2080828 World 0 0.3
## 6 0.5584864 -0.75081900 -1.0479844 World 0 0.3
## 7 -1.2765922 2.08716655 1.4411577 Hello 0 1.3
## 8 -0.5732654 0.01739562 -1.0158475 Hello 0 1.3
## 9 -1.2246126 -1.28630053 0.4119747 World 0 2.3
## 10 -0.4734006 -1.64060553 -0.3810761 Hello 1 1.3
## # ... with 30 more rows
select(td, -starts_with("X"))## $phy
##
## Phylogenetic tree with 40 tips and 39 internal nodes.
##
## Tip labels:
## s1, s2, s3, s4, s5, s6, ...
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [40 x 3]
##
## D1 D2 D3
## <fctr> <int> <dbl>
## 1 World 1 0.3
## 2 Hello 0 2.3
## 3 World 0 2.3
## 4 World 0 0.3
## 5 World 0 0.3
## 6 World 0 0.3
## 7 Hello 0 1.3
## 8 Hello 0 1.3
## 9 World 0 2.3
## 10 Hello 1 1.3
## .. ... ... ...
In many cases, the user may simply want to split apart the treedata object after matching and proceed in their analyses as normal. For example, we could measure phylogenetic signal in our trait X1:
phytools::phylosig(td$phy, getVector(td, X1))## [1] 0.05983494
You can also run it directly on the treedata object using the function treedply:
treedply(td, phytools::phylosig(phy, getVector(td, X1), "K"))## [1] 0.05983494
Or multiple functions at once:
treedply(td, list("K" = phytools::phylosig(phy, getVector(td, X1), "K"),
"lambda" = phytools::phylosig(phy, getVector(td, X1), "lambda"))
)## $K
## [1] 0.05983494
##
## $lambda
## $lambda$lambda
## [1] 7.351765e-05
##
## $lambda$logL
## [1] -48.07617
We can also use the function tdapply which calls the apply function, but allows inclusion of the phylogeny. This can be useful for applying the same function over every column in our dataset. First though, we can use the functions forceNumeric and forceFactor to make sure that every column is of a type that can be analyzed by a function like phenogram or phylosig. These functions to force the traits in the treedata object to be a factor or a continuous data, dropping those that cannot be converted.
tdDiscrete <- forceFactor(td)## Warning in forceFactor(td): Data contain numeric entries, which will be
## converted to factors
## Warning in forceFactor(td): Conversion failed for data columns X1 X2 X3 as
## these data have no shared states. These data will be removed
tdNumeric <- forceNumeric(td)## Warning in forceNumeric(td): Not all data continuous, dropping non-numeric
## data columns: D1
We can further filter out missing data in the trait XNA1.
tdNumeric <- filter(tdNumeric, !is.na(XNA1))Then we can apply a function like phenogram to plot all the data:
par(mfrow=c(2,3))
tdapply(tdNumeric, 2, phytools::phenogram, tree=phy, spread.labels=FALSE, ftype="off")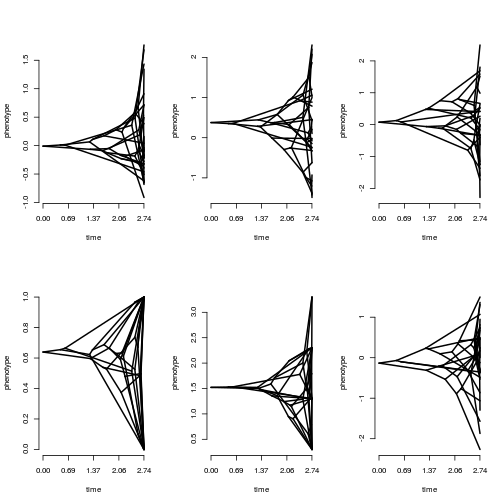
## X1 X2 X3 D2 D3 XNA1
## [1,] NA NA NA NA NA NA
## [2,] NA NA NA NA NA NA
## [3,] NA NA NA NA NA NA
## [4,] NA NA NA NA NA NA
## [5,] NA NA NA NA NA NA
## [6,] NA NA NA NA NA NA
## [7,] NA NA NA NA NA NA
## [8,] NA NA NA NA NA NA
## [9,] NA NA NA NA NA NA
## [10,] NA NA NA NA NA NA
## [11,] NA NA NA NA NA NA
## [12,] NA NA NA NA NA NA
## [13,] NA NA NA NA NA NA
## [14,] NA NA NA NA NA NA
## [15,] NA NA NA NA NA NA
## [16,] NA NA NA NA NA NA
## [17,] NA NA NA NA NA NA
## [18,] NA NA NA NA NA NA
## [19,] NA NA NA NA NA NA
## [20,] NA NA NA NA NA NA
## [21,] NA NA NA NA NA NA
## [22,] NA NA NA NA NA NA
## [23,] NA NA NA NA NA NA
## [24,] NA NA NA NA NA NA
## [25,] NA NA NA NA NA NA
## [26,] NA NA NA NA NA NA
## [27,] NA NA NA NA NA NA
## [28,] NA NA NA NA NA NA
## [29,] NA NA NA NA NA NA
## [30,] NA NA NA NA NA NA
## [31,] NA NA NA NA NA NA
## [32,] NA NA NA NA NA NA
## [33,] NA NA NA NA NA NA
## [34,] NA NA NA NA NA NA
## [35,] NA NA NA NA NA NA
## [36,] NA NA NA NA NA NA
## [37,] NA NA NA NA NA NA
## [38,] NA NA NA NA NA NA
## [39,] NA NA NA NA NA NA
## [40,] NA NA NA NA NA NA
## [41,] NA NA NA NA NA NA
## [42,] NA NA NA NA NA NA
## [43,] NA NA NA NA NA NA
## [44,] NA NA NA NA NA NA
## [45,] NA NA NA NA NA NA
## [46,] NA NA NA NA NA NA
## [47,] NA NA NA NA NA NA
## [48,] NA NA NA NA NA NA
## [49,] NA NA NA NA NA NA
## [50,] NA NA NA NA NA NA
## [51,] NA NA NA NA NA NA
## [52,] NA NA NA NA NA NA
## [53,] NA NA NA NA NA NA
## [54,] NA NA NA NA NA NA
## [55,] NA NA NA NA NA NA
## [56,] NA NA NA NA NA NA
## [57,] NA NA NA NA NA NA
## [58,] NA NA NA NA NA NA
## [59,] NA NA NA NA NA NA
## [60,] NA NA NA NA NA NA
Or you could fit all traits to a BM model and then pull out the sigsq parameter:
fitsBM <- tdapply(tdNumeric, 2, geiger::fitContinuous, phy=phy, model="BM")
sapply(fitsBM, function(x) x$opt$sigsq)## X1 X2 X3 D2 D3 XNA1
## 5.3712587 3.9081367 11.1951598 0.9168851 19.4370364 8.8442750
Perhaps more elegantly, you could use pipes to chain all of these operations together:
td %>% filter(., !is.na(XNA1)) %>% forceNumeric(.) %>% tdapply(., 2, phytools::phylosig, tree=phy)## Warning in forceNumeric(.): Not all data continuous, dropping non-numeric
## data columns: D1
## X1 X2 X3 D2 D3 XNA1
## 0.04615520 0.12933582 0.05012148 0.12608836 0.02233654 0.04680584
You can manipulate the tree as well, using the function treeply, which is meant for simple operations on the tree alone that may or may not change the number of tips. For example, let's use the geiger function rescale.phylo to rescale the branches according to an OU model with an alpha value of 10.
td.OU10 <- treeply(td, geiger::rescale, model="OU", 10)
par(mfrow=c(1,2))
plot(td$phy)
plot(td.OU10$phy)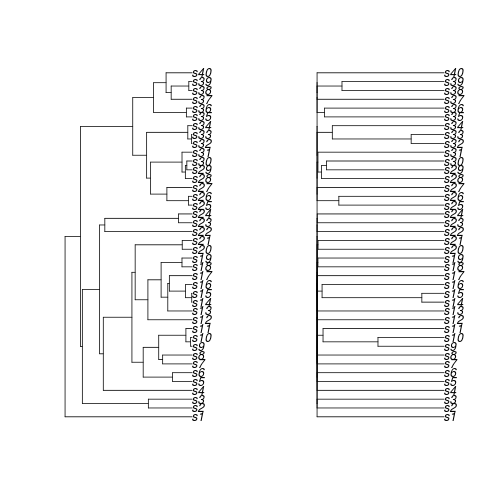
Or we could drop tips from the tree (here we drop tips from 1 to 35).
treeply(td, drop.tip, c(1:35))## $phy
##
## Phylogenetic tree with 5 tips and 4 internal nodes.
##
## Tip labels:
## [1] "s36" "s37" "s38" "s39" "s40"
##
## Rooted; includes branch lengths.
##
## $dat
## Source: local data frame [5 x 7]
##
## X1 X2 X3 D1 D2 D3 XNA1
## <dbl> <dbl> <dbl> <fctr> <int> <dbl> <dbl>
## 1 0.71266631 -0.03472603 1.8031419 Hello 1 1.3 0.7395892
## 2 -0.07356440 0.78763961 -0.3311320 World 0 0.3 -1.0634574
## 3 -0.03763417 2.07524501 -1.6055134 World 1 0.3 0.2462108
## 4 -0.68166048 1.02739244 0.1971934 Hello 0 2.3 -0.2894994
## 5 -0.32427027 1.20790840 0.2631756 World 1 0.3 -2.2648894
One of the cool features about dplyr is that it allows you to group variables and perform analyses independently on different groups with a single command. Currently, treeplyr only supports a single grouping variable at a time, but allows similar functionality. In this example, we will group taxa by the trait D1. You could easily imagine using this for a taxonomic level on the tree.
td.D1 <- group_by(td, D1)What good does this do? Well, we can use summarize to apply functions to specific groups.
summarize(td.D1, mean(X1), sd(X1), mean(X2), sd(X2))## Source: local data frame [2 x 5]
##
## D1 mean(X1) sd(X1) mean(X2) sd(X2)
## <fctr> <dbl> <dbl> <dbl> <dbl>
## 1 Hello 0.09735458 0.9546852 0.10771534 1.089466
## 2 World 0.07318464 0.7174964 -0.01201879 1.231206
But what about if our functions require a phylogeny? Well, we can do that too:
summarise(td.D1, ntips = length(phy$tip.label),
psig.X1 = phytools::phylosig(setNames(X1, phy$tip.label), tree=phy),
psig.X2 = phytools::phylosig(setNames(X2, phy$tip.label), tree=phy))## Source: local data frame [2 x 4]
##
## D1 ntips psig.X1 psig.X2
## <fctr> <int> <dbl> <dbl>
## 1 Hello 17 0.05241561 0.2112923
## 2 World 23 0.29027126 0.3809347
Note both British and American spellings of summarize/summarise work.You might also want to do something like find the total branch length found in different groups of taxa:
summarise(td.D1, ntips = length(phy$tip.label),
totalTL = sum(phy$edge.length), varianceBL = var(phy$edge.length))## Source: local data frame [2 x 4]
##
## D1 ntips totalTL varianceBL
## <fctr> <int> <dbl> <dbl>
## 1 Hello 17 18.23593 0.3182517
## 2 World 23 30.73710 0.5296092
Or you could fit models of trait evolution to different groups in the tree (but note this is a dumb way to build a summary table, as the fitContinuous function is run independently for each parameter).
summarise(td.D1, sigsq = geiger::fitContinuous(phy, setNames(X1, phy$tip.label))$opt$sigsq,
root = geiger::fitContinuous(phy, setNames(X1, phy$tip.label))$opt$z0)## Source: local data frame [2 x 3]
##
## D1 sigsq root
## <fctr> <dbl> <dbl>
## 1 Hello 7.9724838 0.36027556
## 2 World 0.6679094 0.04721332
We can also create a new variable that paints clades according to a particular set of nodes or branches (assuming a postordered tree). by using the function paint_clades.
td.painted <- paint_clades(td, interactive=FALSE, type="nodes", ids=c(75, 66, 54, 48), plot=TRUE)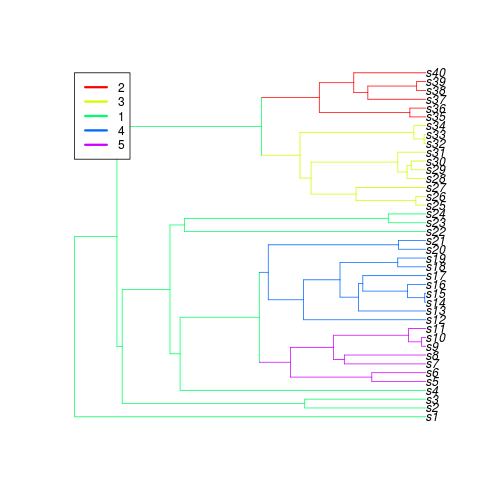
Or alternatively, you can specify which clades you want to group interactively. In the case below, the user selects 4 clades by clicking on the desired branches (i.e. when you run this script,YOU must click on 4 branches of your choosing to move forward!!):
td.painted <- paint_clades(td, 4, interactive=TRUE)Now we group the phylogeny by the clades variable we just defined and calculate summary statistics for each group.
td.painted <- group_by(td.painted, clades)
summarise(td.painted, psig1 = phytools::phylosig(setNames(X1, phy$tip.label), tree=phy),
meanX1 = mean(X1), sdX1 = sd(X1), ntips =length(phy$tip.label))## Source: local data frame [5 x 5]
##
## clades psig1 meanX1 sdX1 ntips
## <dbl> <dbl> <dbl> <dbl> <int>
## 1 1 0.27062158 0.40655789 0.9238538 7
## 2 2 0.27862586 -0.08410896 0.4587212 6
## 3 3 0.03721839 -0.04394916 0.5549763 10
## 4 4 0.12079308 0.34594323 0.9389773 10
## 5 5 0.60748135 -0.28898824 1.0256955 7
Currently, the functions treeply, treedply and tdapply do not work with grouped data frames, and will analyze the entire dataset rather than specified subgroups. This will be added in future versions of treeplyr.