WebappUsage
Once your dataset has finished loading, you should be able to access the web application simply by browsing to http://localhost:5006/nplinker. If everything worked correctly, you will see a page layout similar to the image below:
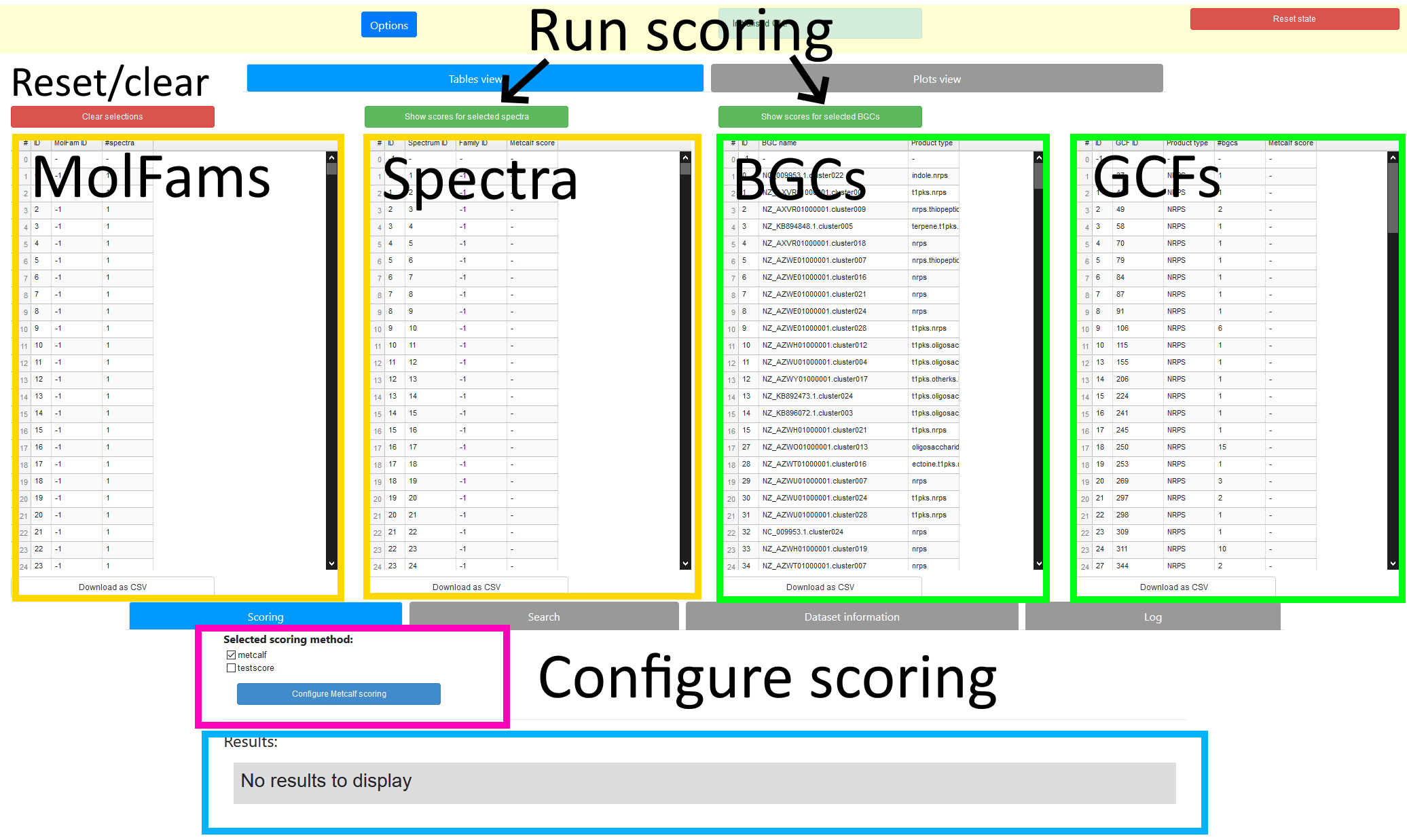
The fundamental feature of the web application interface is the set of 4 tables displayed side-by-side. Each of these tables contains a list of the objects NPLinker was able to extract from the dataset. From left to right, they are: Molecular Families (MFs), spectra, BGCs, and GCFs.
Please be aware that this is not necessarily a complete list of all objects in the dataset! As mentioned in the previous section, any objects with low Metcalf scores are filtered out before this point. The threshold for inclusion is shown above the tables, and can be modified in the configuration file if required. To do this:
- Close any active instance of the webapp by running
docker container rm -f webapp - Open the
nplinker.tomlfile in the shared folder you created previously - Add the following 2 lines to the end of the file to set a new threshold
[webapp]
tables_metcalf_score = 3.0
- Save the file
- Run the webapp again: this will detect the change to the threshold and update the objects listed in the tables accordingly
Another important point concerns the columns shown in the tables. In each case:
- The "#" column is a simple row index
- The "ID" column displays an internal unique ID that NPLinker assigns within each set of objects
- The "MolFam ID", "Spectrum ID" and "GCF ID" columns display identifiers parsed from the dataset. There is no link between the values of the "ID" column and the other columns.
An NPLinker "scoring method" is a generic name for a process which takes in a list of genomics or metabolomics objects and returns a set of links for those objects. The first one implemented by NPLinker is the well-known Metcalf scoring method. In this case, given objects A and B, a link exists between A and B if the Metcalf score for the pair (A, B) exceeds a given threshold. The filtering done by the tables relies on using this method with a relatively low threshold. Once you have selected a set of objects using the tables, you can then perform more strict scoring by running the Metcalf scoring separately on the selected objects. In this case you can easily change the threshold value by clicking the "Configure Metcalf scoring" button.
NPLinker currently includes two other scoring methods: "Rosetta" and "NPClassScore". Rosetta relies on the presence of knownclusterblast .txt files in the "antismash" folder of your dataset and so may not always be usable. NPClassScore relies on predicted chemical compound classes from the MS/MS spectra. Currently, CANOPUS and MolNetEnhancer output can be used for this. CANOPUS can be run from within NPLinker by changing the toml file to include run_canopus = true under the [docker] section, and MolNetEnhancer output can be downloaded from GNPS and stored in a subdirectory called 'molnetenhancer' in the nplinker_data directory. NPClassScore is currently only implemented in the python version and not yet implemented in the webapp. (TODO: explain in more detail somewhere)
Future versions of NPLinker may include other scoring methods. When multiple methods are enabled, they can be combined in any desired combination by ticking/unticking the checkboxes under the "Selected scoring methods" heading.
The goal of this tables-based interface is to make it easy to select a set of one or more objects (e.g. spectra or GCFs) that you wish to find links for and then run a group of one or more scoring methods on them. The generic workflow the app is designed around is:
- Pick one or more objects of the same type you are interested in
- Select the rows corresponding to the object(s) in the appropriate table
- The other tables will now be filtered so that you only see objects that are directly related to the selected object(s). The filtering process is easiest to explain by example. If you select a molecular family, the following filtering steps take place:
- remove any spectra which do not belong to the selected family
- remove any BGCs which do not share strains with the remaining spectra
- remove any GCFs which do not contain any of the remaining BGCs
- A similar process is applied when selecting any of the other object types
- The result is that you will be left with a much smaller subset of the original dataset, which can then be examined in more detail
- After making a selection, click one of the green buttons above the spectra/BGC table. Only one will be enabled depending on whether you selected a metabolomics or a genomics object to start with.
- This will take the set of objects in the corresponding table (BGCs or Spectra) and run one or more NPLinker scoring methods on them, displaying detailed results at the bottom of the page
- The scoring methods used can be controlled and configured by the widgets highlighted in the pink box in Figure 1
- You can then make further selections, adjust the scoring method parameters, or click the red "Clear selections" button to reset the tables to their original state
To give a more concrete example using the example dataset, reset the selections if necessary, and then click on row #10 in the leftmost table. This should be the Molecular Family object with ID 9 and MolFam ID -1 (the "-1" ID indicates it is a singleton family). When you click on this row, you should see the following changes in the user interface:
- The spectra table will be filtered to show only the single spectrum belonging to this particular MolFam
- The BGC table will in turn be filtered to show only the BGCs which share a strain with that spectrum
- The GCF table will in turn be filtered to show only the GCFs which contain those BGCs
- The BGC and GCF tables will be faded slightly and selections on them will be disabled. This is done as an indication that the process of filtering down objects started from the metabolomics side (the opposite will apply if you start filtering on the BGC/GCF tables) There should only be a single spectrum left in the spectra table at this point. Click the green "Show scores for selected spectra" button just above the tableThis will run Metcalf scoring between all the pairs of displayed spectra and GCFs, and display the results below the tables (see image below).
- Click on the blue header that says "12 GCFs linked to Spectrum(id=9, …)" to view the list of GCFs in addition to metadata about the Spectrum itself. Each entry in the GCF list can in turn be expanded to view more information about the individual GCFs
- If you want to change the Metcalf scoring threshold, click the "Configure Metcalf scoring" button and adjust the slider to set the new threshold value. Changes should be reflected in existing results immediately
