Efficient and comprehensive metadata acquisition from NCBI databases (includes SRA).
![]()



NCBImeta is a command-line application that retrieves and organizes metadata from the National Centre for Biotechnology Information (NCBI). While the NCBI web browser experience allows filtered searches, the output does not facilitate inter-record comparison or bulk record retrieval. NCBImeta tackles this issue by creating a local database of NCBI metadata constructed by user-defined search criteria and customizable metadata columns. The output of NCBImeta, optionally a SQLite database or text files, can then be used by computational biologists for applications such as record filtering, project discovery, sample interpretation, or meta-analyses of published work.
- NCBImeta is written in Python 3 and supported on Linux and macOS.
- Dependencies that will be installed are listed in requirements.txt.
- Python Versions:
- 3.7
- 3.8
- 3.9
- Operating Systems:
- Ubuntu
- macOS
Conda is the recommended installation method. To install with pip, gcc is required.
There are three installation options for NCBImeta:
*
mambais strongly recommended overconda!
conda env create -f environment.yaml
conda activate ncbimeta*
gccis required.
pip install ncbimetagit clone https://github.com/ktmeaton/NCBImeta.git
cd NCBImeta
pip install .Test that the installation was successful:
NCBImeta --versionusage: NCBImeta [-h] --config CONFIGPATH [--flat] [--version]
[--email USEREMAIL] [--api USERAPI]
[--force-pause-seconds USERFORCEPAUSESECONDS]
NCBImeta: Efficient and comprehensive metadata retrieval from the NCBI
databases.
optional arguments:
-h, --help show this help message and exit
--config CONFIGPATH Path to the yaml configuration file (ex. config.yaml).
--flat Don't create sub-directories in output directory.
--version show program's version number and exit
--email USEREMAIL User email to override parameter in config file.
--api USERAPI User API key to override parameter in config file.
--force-pause-seconds USERFORCEPAUSESECONDS
FORCE PAUSE SECONDS to override parameter in config
file.
--quiet Suppress logging of each record to the console.
Download the NCBImeta github repository to get access to the example configuration files:
git clone https://github.com/ktmeaton/NCBImeta.git
cd NCBImetaDownload a selection of genomic metadata pertaining to the plague pathogen Yersinia pestis.
NCBImeta --flat --config test/test.yaml(Note: The 'quick' start config file forces slow downloads to accommodate users with slow internet. For faster record retrieval, please see the config file docs to start editing config files.)
Example output of the command-line interface (v0.6.1):

NCBImetaAnnotate \
--database test/test.sqlite \
--annotfile test/test_annot.txt \
--table BioSampleNote that the first column of your annotation file MUST be a column that is unique to each record. An Accession number or ID is highly recommended. The column headers in your annotation file must also exactly match the names of your columns in the database.
NCBImetaAnnotate by default replaces the existing annotation with the data in your custom metadata file. Alternatively, the flag --concatenate can be specified. This will concatenate your custom metadata with the pre-existing value in the database cell (separated by a semi-colon).
NCBImetaAnnotate \
--database test/test.sqlite \
--annotfile test/test_annot.txt \
--table BioSample \
--concatenateNCBImetaJoin \
--database test/test.sqlite \
--final Master \
--anchor BioSample \
--accessory "BioProject Assembly SRA Nucleotide" \
--unique "BioSampleAccession BioSampleAccessionSecondary BioSampleBioProjectAccession"The rows of the output "Master" table will be from the anchor table "BioSample", with additional columns added in from the accessory tables "BioProject", "Assembly", "SRA", and "Nucleotide". Unique accession numbers for BioSample (both primary and secondary) and BioProject allow this join to be unambiguous.
NCBImetaExport \
--database test/test.sqlite \
--outputdir testEach table within the database will be exported to its own tab-separated .txt file in the specified output directory.
- Explore your database text files using a spreadsheet viewer (Microsoft Excel, Google Sheets, etc.)
- Browse your SQLite database using DB Browser for SQLite (https://sqlitebrowser.org/)
- Use the columns with FTP links to download your data files of interest.
Example database output (a subset of the BioSample table)
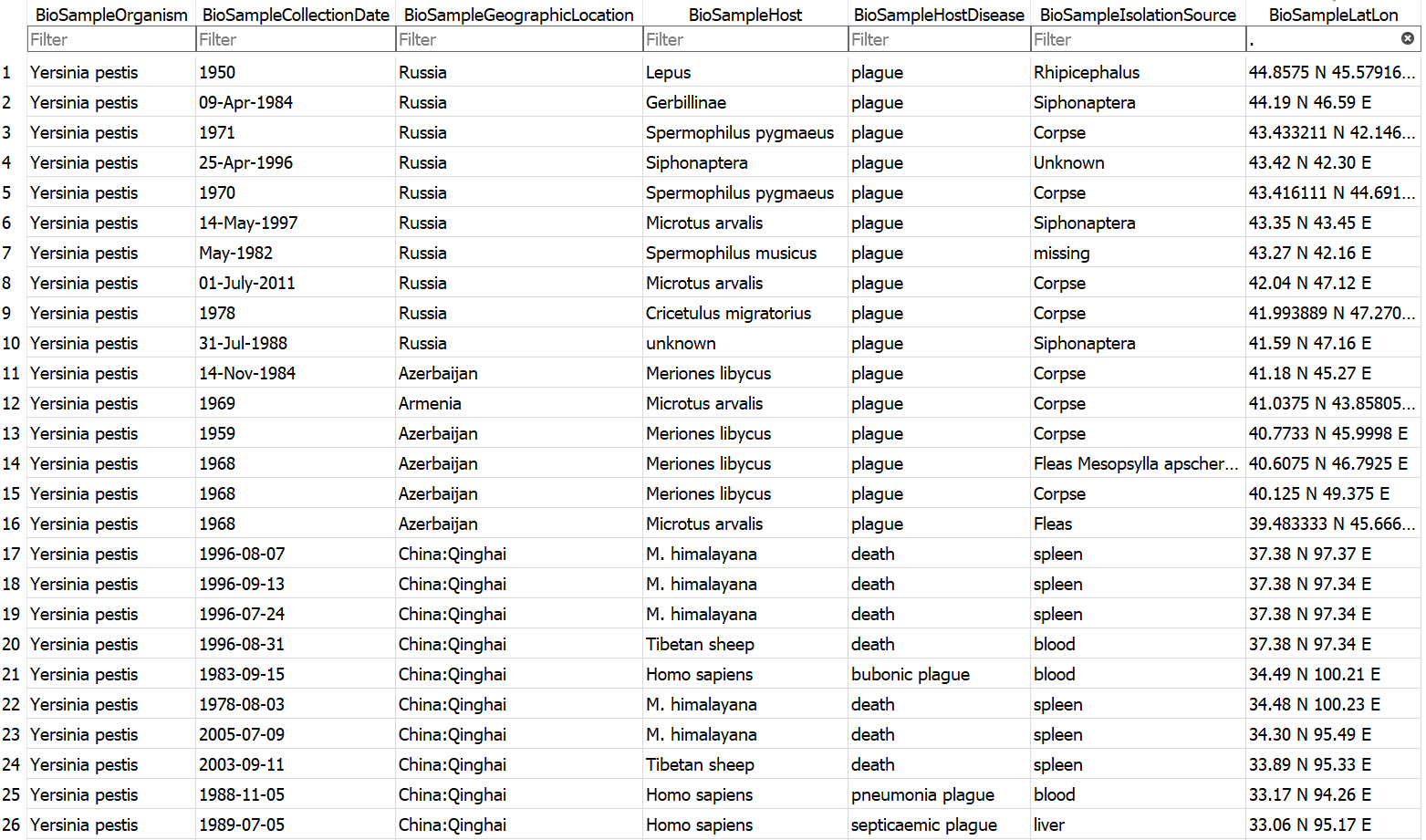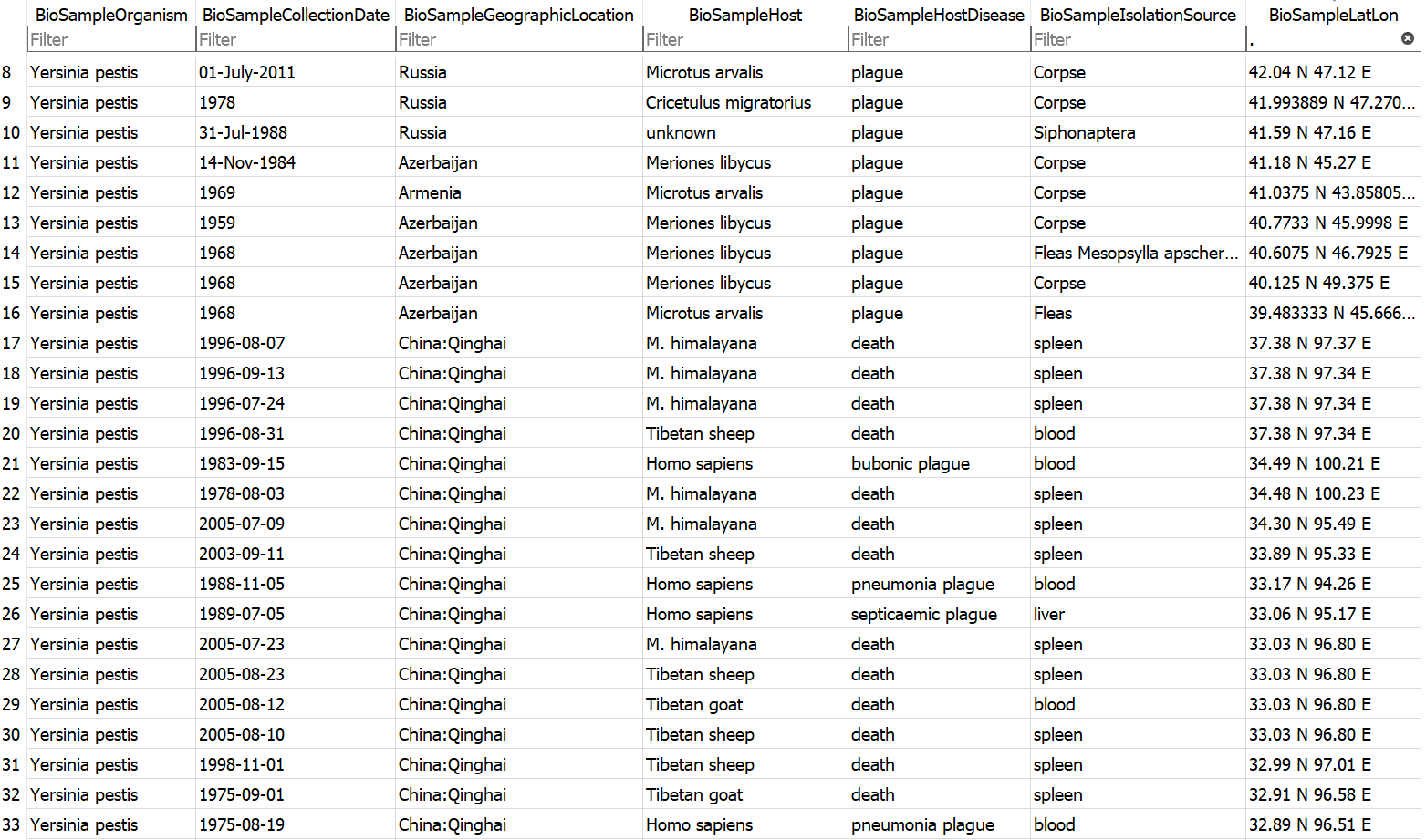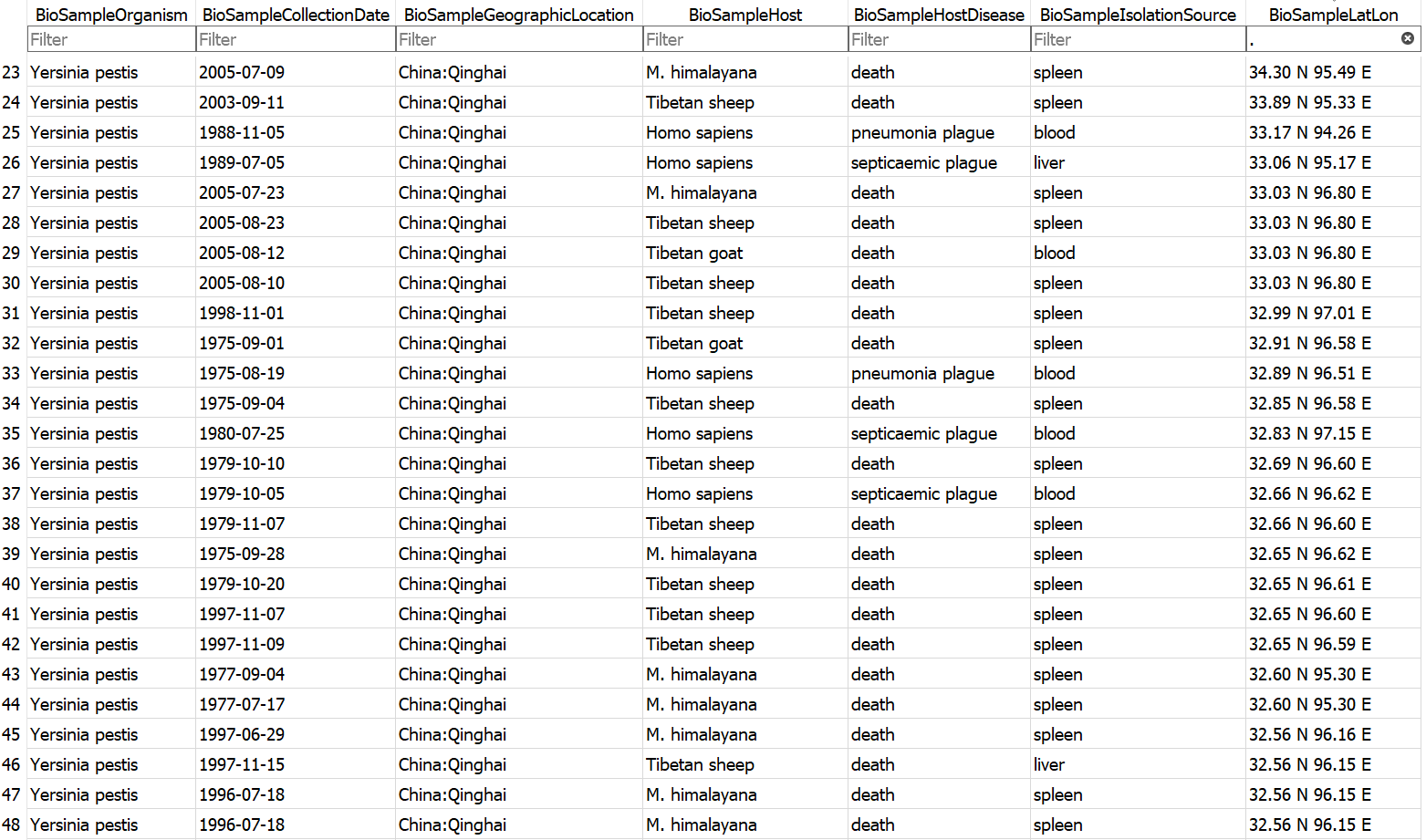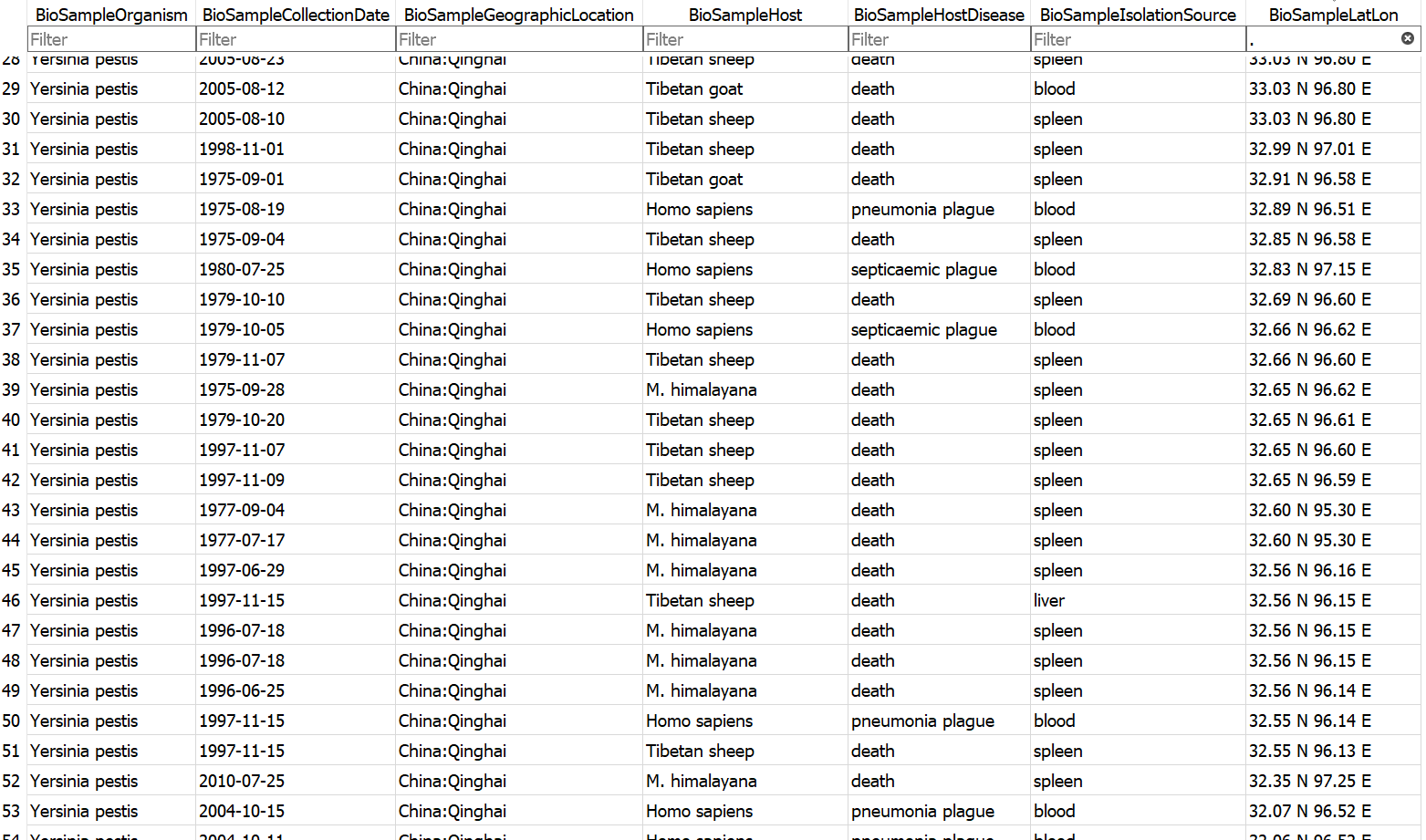
- Assembly
- BioProject
- BioSample
- Nucleotide
- SRA
- Pubmed
- Project "Read The Docs": Documentation Overhaul - PLANNED
- Project v0.8.3 - "Update Dependencies": Bugfixes for Installation - RELEASED
- Project v0.8.2 - "Annotate Simplicity": Simplify the Annotate Command - RELEASED
To get started with customizing the search terms, database, and metadata fields, please read:
Please submit your questions, suggestions, and bug reports to the Issue Tracker.
Please do not hesitate to post any manner of curiosity in the "Issues" tracker :) User-feedback and ideas are the most valuable resource for emerging software.
GitHub not your style? Join the NCBImeta Slack Group to see release alerts, chat with other users, and get insider perspective on development.
Want to add features and fix bugs? Check out the Contributor's Guide for suggestions on getting started.
Eaton, K. (2020). NCBImeta: efficient and comprehensive metadata retrieval from NCBI databases. Journal of Open Source Software, 5(46), 1990, https://doi.org/10.21105/joss.01990
Author: Katherine Eaton ([email protected])
Those who have filed issues, pull-requests, and participated in discussions.

{kind=link}