BLENDER is a companion program to the DISCOVER-Seq assay to identify off-target CRISPR/Cas editing sites from MRE11 ChIP-Seq experiments described in "Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq." BLENDER takes aligned bamfiles from the MRE11 IP experiment and (optionally) a control bamfile and identifies locations with stacks of reads at putative cutsites. PAM sequences can be provided by the user as well as a guide sequence. BLENDER makes use of the ENCODE ChIP-Seq Blacklists for human and mouse which are lists of regions in the genome that have artifactual large numbers of reads. These lists and the control bam plus PAM sequences and the guide are used to filter out false positives. BLENDER runs on mouse mm10 and human hg38 genomes (blacklists coordinates are for these genomes).

As of March 2023, there are two versions of BLENDER.
- "New" BLENDER (aka "BLENDER2") uses the same logic as classic BLENDER, but has been rewritten to be much faster and easier to develop against. BLENDER2 will replace BLENDER for all future uses of DISCOVER-seq. Stay tuned for several updates to BLENDER2!
- "Classic" BLENDER (aka "BLENDER") is the original software reported in Wienert and Wyman et al Science 2019. It is battle-tested but slow. A single run can take 1-2 days on a single CPU, depending on sequencing depth. It is also very hard to develop against, and so new features have lagged behind. BLENDER is provided so that one can compare to historical datasets/results.
BLENDER has a driver script run_blender.sh that takes several argmuments and runs identification of putative hits, filtering of those hits, and then creates an SVG of the aligned hits. Files are stored in the output directory given as a parameter to the bash script. Alternatively, each step can be run separately.
sh run_blender2.sh <path to reference genome> \
<path to IP bamfile> \
<path to control bamfile> \
<guide sequence> <output directory> ["options"]
sh run_blender.sh <path to reference genome> \
<path to IP bamfile> \
<path to control bamfile> \
<guide sequence> <output directory> ["options"]
These will run blender with option c set to 3 (details below), this means the program will run quickly, but may miss some very sparsely covered off target sites. I recommend running it initially with c set to 3 (the default) and then running again with c set to 2. Classic blender may take several days to run, but new blender should complete relatively quickly.
There are two major versions of blender:
- blender2.py: This uses the same logic as blender.pl, but is rewritten to be more flexible and have a much faster backend. It takes 5-10 minutes for a typical run to complete. It finds the same sites as blender.pl, but with very slight changes to the score. The Corn Lab has switched to using blender2.py, and this version will be the one developed against for all future versions.
- blender.pl: This version was originally published in Wienert & Wyman et al Science 2019. It is very slow, but has been extensively benchmarked. A single-CPU run can take as long as 1 day to complete and the newer multi-CPU mode can reduce this to 1 hour or so if you have many CPUs. It only works with blunt-cutting Cas enzymes that have a cutsite and PAM location identical to SpyCas9.
Faster, more flexible, less tested
pysam https://pysam.readthedocs.io/en/latest/installation.html (best installed via conda) pandas https://pandas.pydata.org/pandas-docs/stable/getting_started/install.html numpy https://numpy.org/install/
python blender2.py -f <experimental bamfile> -c <control bamfile> -n <nuclease name> -g <guide sequence> -r <reference genome> -b hg38.encode-blacklist.v2.bed -o unfiltered_output.txt
python blender2.py -f <experimental bamfile> -c <control bamfile> -n <nuclease name> -g <guide sequence> -r <reference genome> -b hg38.encode-blacklist.v2.bed --filter -o output.txt
python blender2_mp.py -f <experimental bamfile> -c <control bamfile> -n <nuclease name> -g <guide sequence> -r <reference genome> -b hg38.encode-blacklist.v2.bed --filter -o output.txt -j <number parallel processes>
BLENDER can be run with or without filtering based on number of mismatches and score. We have so far never found a false-negative that was inappropriately filtered out. But we provide the option of outputting unfiltered, raw results just in case you're curious about every single possible hit.
Note that blender does not require the separate filter.pl used by blender.pl

-f --file Experimental bamfile. This is the aligned bamfile for the MRE11 pulldown of ChIP-Seq of a Cas9 edited sample. BLENDER will extract the reference sequence fromthis file for use in the analysis. I typically use BWA for alignment, but bowtie2 can be used as well. BLENDER has not been tested with bamfiles from other aligners. Required.
-o --output Output file Required
-c --control Control bamfile. This is a ChIP-Seq for MRE11 pulldown from either unedited cells or cells that have been edited with a non-targeting gRNA. If there are greater than 10 reads in the control sample, the hit in the edited sample is filtered out.
-n --nuclease Nuclease to search for. Current possibilities are: SpyCas9, SaCas9, LbCas12a, AsCas12a. Required.
-g --guide Guide sequence. Provided 5'-> 3' without the PAM sequence. Required.
-r --reference Reference genome in FASTA format. Must be pre-indexed with faidx so that an accompanying *.fai is found in the same directory as the FASTA-formatted genome. Required.
-b --blacklist Blacklist to use for filtering hits, e.g. from ENCODE (BED format). Very strongly recommended
--filter Filter results by score and mismatches (previously accomplished by separate filter.pl script) Recommended to clean up noise
-w --window_size Override nuclease-default window size for score summing. Cas9 default = 5, Cas12 default = 10'
-p --pams Override nuclease default PAMs with a space-separated list. Does not include the degenerate base. e.g. -p GG AG
-t --threshold Threshold for number of read ends exactly at a putative cut site (default 3). Note that this was formerly option -c in blender.pl, but is now -t to avoid confusion with the control BAM file!
-s --score_min Minimum aggregated score across a 5-base window around the cutsite to consider a hit (default 3)
-m --max_mismatches Maximum number of mismatches to allow to the guide sequence (default 8)
-q --mapq Theshold MAPQ value for reads to be considered (default 20)
--verbose This flag will turn on output of filtered out candidates while running. These sites are not written to the output file, but a log can be captured for troubleshooting.
This is the same as blender2.py, but has an extra option to allow parallel processing. Note that running this with just one core is actually slower than blender2.py. But running blender2_mp.py over many cores in parallel for each chromosome can give a speed increase of up to 6x.
-j --nproc Number of parallel processes to start (default 2). Parallelizes per chromosome, and so maxes out at the number of chromosomes in your bamfile or reference fasta.
Older, much slower, more tested
Parallel::ForkManager https://metacpan.org/pod/Parallel::ForkManager
sambamba https://lomereiter.github.io/sambamba/
samtools http://www.htslib.org/
perl blender.pl [options] <reference genome> <guide sequence> <IP bamfile> <control bamfile> > unfiltered_output.txt
perl blender.pl [options] <reference genome> <guide sequence> <IP bamfile> <control bamfile> | perl filter.pl > output.txt
perl blender.pl [options] <reference genome> <guide sequence> <IP bamfile> <control bamfile> | perl filter_pool.pl > pooled_output.txt
BLENDER can be run with or without being piped through the filtering script. There are two filtering scripts provided; the standard filter.pl script that implements the standard scoring scheme, and the filter_pool.pl script that implements the more stringent scoring scheme for pooled samples.
reference genome Path to reference genome. If reference has "mm10" in it, then the mouse blacklist coordinates will be used. Otherwise, human is assumed and the hg38 blacklist coordinates will be used.
guide sequence Guide sequence should be provided 5'-> 3' without the PAM sequence.
IP bamfile This is the aligned bamfile for the MRE11 pulldown of ChIP-Seq of a Cas9 edited sample. BLENDER will extract the reference sequence fromthis file for use in the analysis. I typically use BWA for alignment, but bowtie2 can be used as well. BLENDER has not been tested with bamfiles from other aligners.
control bamfile This is a ChIP-Seq for MRE11 pulldown from either unedited cells or cells that have been edited with a non-targeting gRNA. If there are greater than 10 reads in the control sample, the hit in the edited sample is filtered out.
output directory
-p List of 2 nucleotide PAM sequences, separated by commas, in quotes. The default is "GG,AG".
-c Cutoff threshold for number of read ends at a putative cut site. Default is 3. For maximum sensitivity, this can be set to 2 and the filtering scheme applied. BEWARE that this dramatically slows down running time. It can
increase runtime from ~30min to 24hrs, depending on the guide.
-j Number of threads to use (default 1, ideally equal to or higher than the number of chromosomes in the target genome))
--verbose This flag will turn on output of filtered out candidates while running if filtered out for more than maximum mismatches (8) in the guide sequence, or the hit occurs in a blacklist region or it is in a very deep region and
thus likely an artifact.
blender.pl and blender2.py have similar output formats.
Chr:Start-End Genomic coordinates of the putative guide
Cutsite Where the cutsite is within the guide
Discoscore Score given to the hit. Essentially summing a window of read ends around the cut site
Cutsite Ends This is the number of read ends that pile up at the cutsite. When you set the 'c' parameter, it is this value that is set.
Strand/PAM
Guide sequence
Mismatches
norm_discoscore normalized Discoscore across all unfiltered hits, ranging from 0-1. only in blender2
z_discoscore Z-score of the Disoscore vs all other Discoscores at all other blunt reads in the entire genome. only in blender2
The automated bash scripts output unfiltered_blender_hits.txt, filtered_blender_hits.txt and blender_hits.svg to the output directory provided by the user. This raw unfiltered output can be used for exploring bamfiles to assess whether adjustments might be needed for the scoring scheme.
You can use draw_blender_fig.py to create an svg image of the hits from either blender or blender2.
-i, --infile blender output file to draw
-o, --outfile output filename
-g, --guide Guide RNA sequence (5' to 3')
-p, --pam PAM to output (5' to 3')
-l, --location PAM location (5' or 3' of the guide RNA). Default 3
-t, --title Optional title to add to the plot"
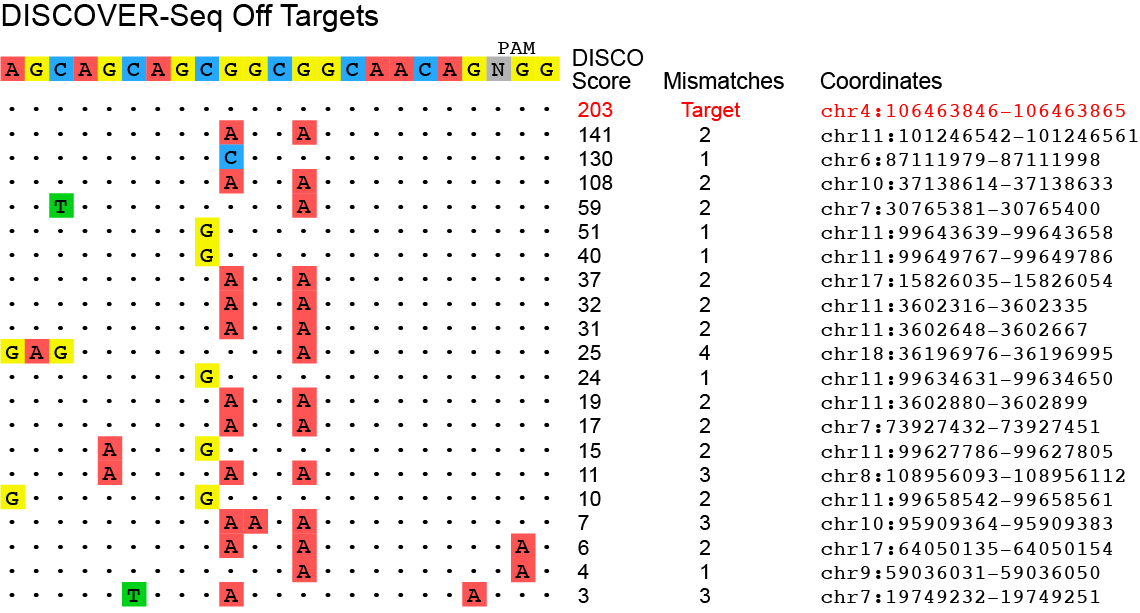
Demo data is included in the DEMO file. The DEMO directory contains data for running BLENDER on a single chromosome (chr19) for DISCOVER-Seq from MRE11 ChIP-Seq of the VEGFA site 2 guide in K562 cells.
Bam files for the IP file and the control file (BFP ChIP) are provided in the bwa directory and expected output can be found in the expected_output directory. Chromosome 19 has three off-target hits.
To run BLENDER on the demo data, use the following command in the main blender directory:
sh run_blender.sh <path/to/reference/genome> \
DEMO/bwa/BW43_VEFGA.chr19.bam \
DEMO/bwa/BW44_BFP_control.chr19.bam \
GACCCCCTCCACCCCGCCTC DEMO/blender
sh run_blender2.sh <path/to/reference/genome> \
DEMO/bwa/BW43_VEFGA.chr19.bam \
DEMO/bwa/BW44_BFP_control.chr19.bam \
GACCCCCTCCACCCCGCCTC DEMO/blender
For BLENDER, this will take approximately a minute or less to run. Runtime for BLENDER2 is about 2 seconds. The three output files can be found in the DEMO/blender directory. Because the bam file is just for chromosome 19, the output figure shows just the 3 hits found on chromosome 19 (image below is for BLENDER, with same sites but slightly different score for BLENDER2)

The fastq files to run this data on all chromosomes can be found in the NCBI Short Read Archive with BioProject Accession PRJNA509652. The links to the fastq files are (click on "Data Access" tab to get fastq download):
VEGFA IP Fastqs (BW43): https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR8550675
BFP control fastqs (BW44): https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR8550676
*Wienert, B., *Wyman, S. K., Richardson, C. D., Yeh, C. D., Akcakaya, P., Porritt, M. J., Morlock, M., Vu, J. T., Kazane, K. R., Watry, H. L., Judge, L. M., Conklin, B. R., Maresca, M. and Corn, J. E. (2019). Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. *contributed equally