Home
This is an implementation of an efficient and simple model to simulate high-resolution atomic force microscopy (AFM), scanning probe microscopy (STM) and inelastic tunnelling microscopy (IETS) images using classical forcefields. The main principle is illustrated in the following image:
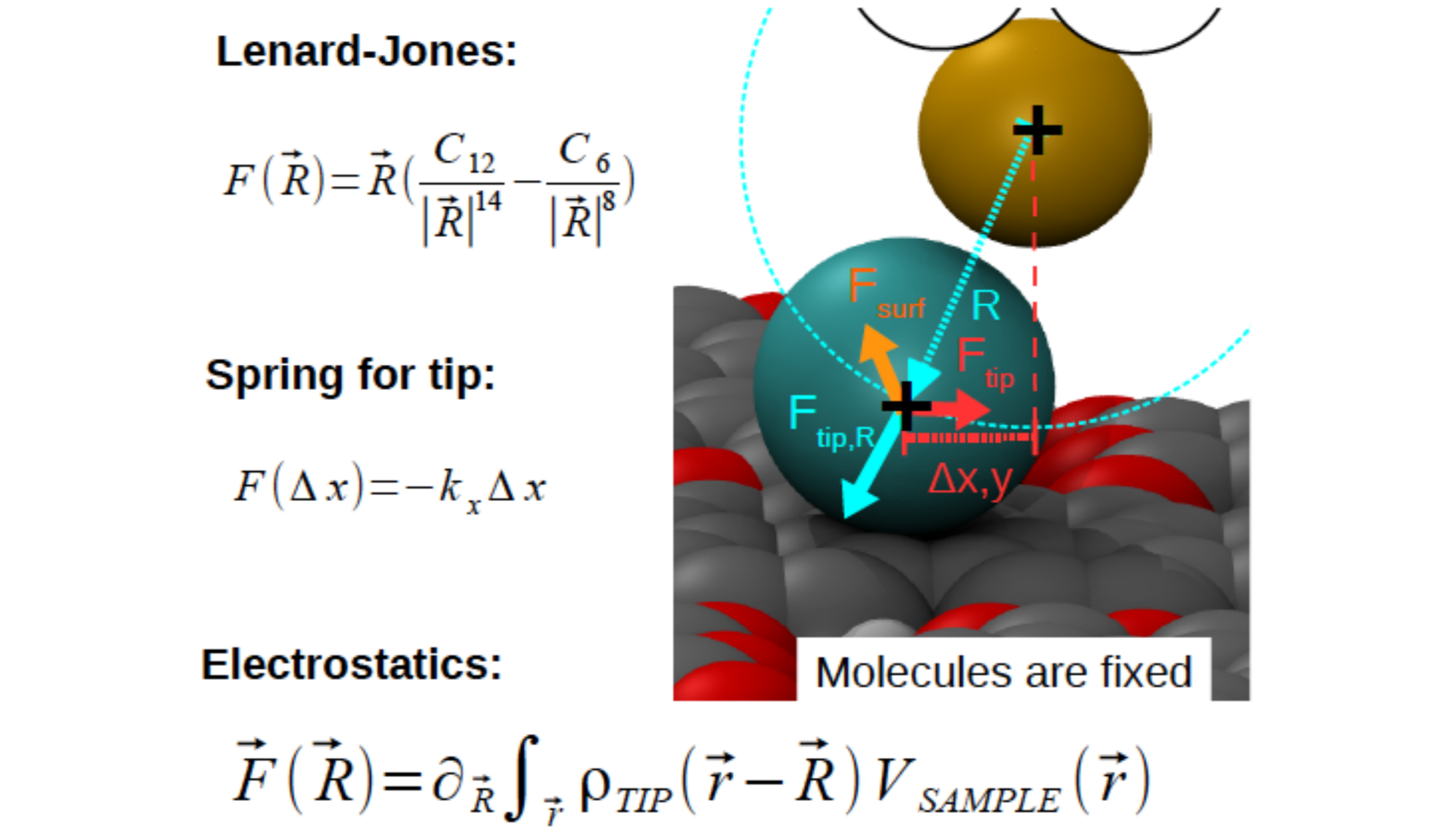
The schematics illustrate all the forces acting on the relaxing probe particle (PP - blue ball), which represents the last atom of the flexible tip apex. Those forces are i) spring forces - representing the forces of the tip and keeping the PP below the last atom of the metallic tip-base (yellow ball); ii) Pauli repulsion and van der Waals attraction between the PP and fixed atoms of the substrate approximated by the Lennard-Jones potential; iii) Electrostatic forces between sample (calculated via DFT) and (monopole/dipole/quadrupole) charged PP (if PP/flexible tip apex is charged). A detailed description of the forces and differences between the original paper and the current implementation are described here
The code has a form of Python module which imports C/C++ library using ctypes. This way it should be very friendly to any further development and integration to other projects.
Check here for the installation instructions.
For the minimal input without any electrostatics only atomic positions are needed:
- They can be provided in a bas or xyz format in and
*.xyzfile. . You can also add (e.g. Mulliken) charges as a 5th column to this file. See the forcefield section for more details. Also don't forget to specify the cell in theparams.inifile, or change the cell parts of yourparams.inifile. - Another option is to use FHI-AIMS geometry format
*.in. The cell can be read directly from the geometry file if specified there. - Currently also the POSCAR or CONTCAR from VASP can be used.
If you have the possibility to calculate the electrostatic (= Hartree) potential using a DFT code, this should give you more precise results. At the moment 4 codes are supported: Fireball, VASP, FHI-AIMS and CP2K. (Using other codes shouldn't be a big problem, but beware of different units. Please write us if you are unsure, or you are running into problems.) The files can be used to provide everything - information about electrostatics, geometry, and cell. Look at the forcefield section for how to run the calculations using these. With these 4 codes, two input file types should be used:
-
*.xsf-- from Fireball or VASP (use v2xsf to transferLOCPOTintoLOCPOT.xsf). Potential is in eV (same units as ppafm uses). -
*.cube-- from FHI-AIMS or CP2K. Potential in Hartree. ppafm uses internal rescaling. - Please do not mix these workflow paths, otherwise, you can encounter problems with units.
We have prepared a few examples which should run out-of-the-box under Linux just by running the run.sh bash script. This includes
- A very simple and fast calculation of Graphene with nitrogen defects using a point-charge model of electrostatics.
- A bit more sophisticated calculation of monolayer of PTCDA molecule on Ag111 with DFT Hartree potential as input. The Hartree potential in
LOCPOT.xsfis downloaded automatically bywgetfrom our hosting. (Usually doesn't work on OSX, it is better to download the file manually and updaterun.shaccordingly). There are two examples: first of them is using monopole or quadrupole charge on the probe particle (PP). The differences between monopole and quadrupole charge is illustrated in figure from Supplementary Information of Nat. Commun. 7, 11560 (2016):
- IETS example recalculating the theoretical data without any electrostatics, published in PRL 113, 226101 (2014).
As you can see from the content of the directories, 2 input files are required:
-
params.iniwhich contains input parameters of the simulation. A dictionary of all parameters can be found on the wiki page about setting simulation parameters. -
*.xyzwhich contains input atomistic geometry (first column atom type, followed by 3 columns of x,y,z Cartesian coordinates of the atom, followed by an optional column of atomic charges) ... see example- In more sophisticated calculations you can use the Hartree potential from a DFT calculation instead of atomic charges. In this situation also the atomic geometry can be read from the
*.xsfor*.cubefile. An example of the first of them can be seen while running the example inhttps://github.com/Probe-Particle/ppafm/tree/master/examples/PTCDA_Hartree - Atomic geometry and parameters of the cell can be also read from the
geometry.inFHI-AIMS file.
- In more sophisticated calculations you can use the Hartree potential from a DFT calculation instead of atomic charges. In this situation also the atomic geometry can be read from the
To run the calculations directly and see the results instantaneously you can use Graphical User Interface (GUI) through ppafm-gui, which implicitly calculates everything on a GPU. The GUI is described here.
To have a larger amount of options or for a better understanding of the inner process you can see in run.sh the df calculation is always composed of 3 independent steps:
This is done by running the script (all the ppafm scripts are explained here):
ppafm-generate-ljff -i input.xyz
ppafm-generate-elff-point-charges -i input.xyz
if you use the point-charge electrostatics model and geometry in input.xyz or by:
ppafm-generate-ljff -i LOCPOT.xsf
ppafm-generate-elff -i LOCPOT.xsf
if you have a Hartree potential and a geometry stored in LOCPOT.xsf. The electrostatic forcefield is necessary only for a calculations with charged tip and in both cases it is created during running the second command. The computation of the electrostatic force is mainly explained in the Supplementary Material of PRL 113, 226101 (2014).
The output FFLJ_?.xsf are the x,y,z component of the Lennard-Jones force field in units [eV/Å] for the positions of PP over the sample on a grid defined by the sampling of input LOCPOT.xsf or defined by gridA, gridB, gridC, gridN parameters in params.ini.
The FFel_?.xsf contain the x,y,z components of the electrostatic force field in [V/Å] (scaling by the PP charge Q is done only in relaxed scan procedure).
where the position of the PP is relaxed in order to minimize its total energy, i.e. to find a balance of forces between forces from the tip and the sample. The sample force is found by an interpolation of the total force-field obtained as Ftot = FFLJ + Q*FFel. This is done by executing the script:
ppafm-relaxed-scan -k 0.5 --qrange -0.05 0.0 2 --pos
where -k denotes tip stiffness in [N/m]; --qrange range of charges on PP, and --pos indicates that, in addition to the force, also relaxed positions of PP should be exported. The results for different stiffnesses and PP charges are stored to separate directories.
where we just read the output OutFz.xsf and PPpos_?.xsf and generate a set of images from it. Here, we also convert the vertical force Fz to frequency shift df for given (peak-to-peak) oscillation amplitudes set by -a or --arange and given parameters of the cantilever kCantilever, f0Cantilever (default parameters are for a standard Qplus sensor). This is done by executing the script:
ppafm-plot-results -k 0.5 --qrange -0.05 0.0 2 --arange 0.5 2.0 2 --pos --df
Flag --df enables plotting of the df_???.png images with calculated df data, while --pos plots the positions of PP during the simulated scan into xy__???.png
This is the only part of the procedure where dependence on matplotlib is required. Instead, you can also use other visualization tools:
- By adding the --save_df flag you can save your df results in the df.xsf file, which you can visualize e.g. via VESTA.
- Using the --WSxM flag you will print each height slice into a df_???.xyz file, readable e.g. by the WSxM visualization tools.
Further outputs
The z-component of the force acting on the tip can be plotted into Fz_???.png files by using the --Fz flag. (Second electrostatics is not taken into account in the plotted force!)
The IETS signal (as calculated in PRL 113, 226101 (2014), that means using variation of CO frustrated translation peak energy approximation, see PRL 119, 166001 (2017) for more details) will be calculated and plotted via applying --iets=M,V,W flag, where: M - PP mass [a.u.]; V - bias offset [eV]; W - peak width [eV].
Both of these quantities can be printed into *.xyz files by means of the --WSxM flag.
Optional flags:
-
--cbar: adds a color bar with absolute numbers to the*.pngimages. -
--atoms: adds a position of atoms saved ininput_plot.xyzinto the plotted*.pngimages. -
--bonds: adds also lines in between close-by atoms ... -
--no_int: allows for plotting of images without any interpolation between pixels
You can also speed up the calculations and spare approx. 1/2 of your disc memory by using --npy behind all the scripts. It will save all your intermediate data into "machine-readable" *.npy files, which on the other hand are inconvenient for debugging.
There are also other flags, that are connected with other developments. For example, see KPFM approximations here.
We strongly recommend keeping all the parameters for the calculations (mainly multipole = tip, sigma, PBC and the second multipole = tip_base) in the params.ini instead of using flags.
Any line in the params.ini file can be commented on by using # (ideally with space).
Multipole (dipole, quadrupole) charge is not multipole moment. Dipole moment can be calculated as charge * sigma [e*Å]; quadrupole moment is charge * sigmaˆ2 [e*ň2].
CO quadrupole charge varies from -0.05 to -0.30 (using sigma = 0.71 or 0.7) depending on the experiment.
Stiffness of 0.24-0.25 N/m (stiffness 0.24 0.24 20.0) is mostly used for CO-tips (Nat. Commun. 7, 11560 (2016)).
PBC affects only calculations of LJ potential. The code is always periodic from the nature of its design: PBC False means nPBC 0 0 0 ; PBC True means read nPBC parameter.
nPBC affects only calculations of LJ potential -- how many cells around the original one are used for LJ force-field calculation. The last number plays no role. The total amount of cells is given by (nPBC(x).2+1).(nPBC(y).2+1) and the final geometry is always centered in the original geometry as given in the input file. nPBC 0 0 0 means only the original cell; nPBC 1 1 1 3x3 cells around the original one and so on. See the image below for a graphical representation.

gridN can be assigned automatically (commented or missing in params.ini) with a division of 0.1 A.
The scanning height is referred to the position of the last atom of the metal tip-base. The equilibrium position of the PP is then by r0Probe-z component lower. (4Å is default; e.g. in Nano Lett. 16, pp 1974–1980 (2016) and ACS Nano 12, 5274−5283 (2018) 3Å were used.)
If in IETS you get many negative frequencies, try to change the scanning grid or gridN. There are always a few negative energies, especially at really close distances.
Cell definitions
The code expects, that your system is in the cell starting in 0,0,0, given by the 3 lattice vectors - each of them is a row: 1st is gridA; 2nd gridB and the last one is gridC (normally 0. 0. height of cell ) - basically the same strategy as in ASE:
- For example, if your system has the centre of mass at 0,0,0 you can run into problems (sometimes problems with FHI-aims).
- Or if your geometry is below 0 in z then the atoms are not captured properly, regardless of stated
PBCornPBC. - it is also good practice to leave enough space above the top-most atoms in the z direction: 15 Angstrom is pretty safe.
- which leads to another good practice to shift the system to a lower part of your cell.
If you are using Hartree potential (xsf or cube files); Then you do not need to care about these - the points of the grid are the same as in those files with potential.
- If you want better (more dense) spacing or larger cell, then you need to redo your DFT calculations with adjusted parameters.
If not -- you have to specify: gridA, gridB and gridC by hand (default values do not make any sense):
- if you have a system that is periodic in x and y (surface) then adjust
gridAandgridBwith the exact lattice vector;gridCneeds to have enough space about the atoms (as mentioned above).
Scan definitions
 The estimation of appropriate scan height for forces. Be aware that for the frequency shift we need to account for the oscillation, so this height corresponds to the closest point of the oscillation. You can say that the real scanning distance is the closest point of the oscillation + A/2, where A (peak-to-peak amplitude), when describing the tip-height. However, from the computational point of view it makes more sense to refer to the closest point of oscillation in description, while clearly stating it. Finally, the real tip-sample distance is unknown, as any parametrization we provide, cannot ensure the exact match of short-range F(z) forces. This is even more pronounced, when the long-range forces are in the experiment.
The estimation of appropriate scan height for forces. Be aware that for the frequency shift we need to account for the oscillation, so this height corresponds to the closest point of the oscillation. You can say that the real scanning distance is the closest point of the oscillation + A/2, where A (peak-to-peak amplitude), when describing the tip-height. However, from the computational point of view it makes more sense to refer to the closest point of oscillation in description, while clearly stating it. Finally, the real tip-sample distance is unknown, as any parametrization we provide, cannot ensure the exact match of short-range F(z) forces. This is even more pronounced, when the long-range forces are in the experiment.
Double Electrostatics moved here

Example of a simulated (right) and experimental (left) high-resolution AFM image of a self-assembled monolayer of PTCDA molecules on Ag(111) surface with a Xe tip (at liquid helium temperature with collibry sensor). A positive monopole of 0.3e was used for the simulations
- Probe-Particle Model (in capital letters) - the name of the simulation model/method; Note: in some publications it could be referred to as Probe Particle Model. We will try to stick to the proposed naming in the future.
- probe particle - the simulation particle (e.g. CO or Xe); Note: in some publications it could be referred to as probe-particle or even Probe-Particle. We will try to stick to the proposed naming in the future.
- PP - abbreviation of probe particle
- ppafm - the name of this Python package as well as the general term for an AFM simulation done using the Probe-Particle Model.
The code considers basically three types of interaction between the tip ( PP particle) and the sample.
- Pauli-repulsion ( currently approximated by repulsive part of Lennard-Jones potential
r^-12) - van der Waals attraction ( currently approximated by attractive part of Lennard-Jones potential
r^-6) - Electrostatic ( currently can be computed as Coulomb pairwise interaction between the PP and point charges in centres of atoms of the sample, or by reading the electrostatic force-field obtained by a derivative of sample Hartree potential as described in supplementary of PRL 113, 226101 (2014).
The computation of images is divided into two parts:
- Precompute vector force field ( Fx(x,y,z), Fy(x,y,z), Fz(x,y,z) ) over the sample and store it on a 3D-grid. ( see
getLenardJonesFFandgetCoulombFFfunctions for more details ). After individual components of the force field are sampled ( i.e. Lennard-Jones and electrostatic ) they are summed up to form one single total effective force field in which the PP moves. - Relax the PP attached to the tip under the influence of the total effective force field. The relaxation of the PP is done using the "Fast Inertial Relaxation Engine" (FIRE) algorithm ( see.
PRL 97, 170201 (2006) implemented in
FIRE:move()function. In each step of the relaxation, the force field on the grid is interpolated in theinterpolate3DvecWrapfunction, considering periodic boundary conditions. From Python the relaxation is called asrelaxTipStrokefunction providing a 1D-array of tip positions, and obtaining back 1D-array of PP position after the relaxation and of force between tip PP and sample at that position. The lateral scan in (x,y) to obtain a stack of images is done by callingrelaxTipStrokeat different (x,y) positions of the tip, where each call ofrelaxTipStrokedoes one approach along the z-direction.
This splitting of the computation (first sampling of the force field on the grid, and then relaxation using interpolation) has several advantages over the straightforward computation of interaction on the fly during the relaxation process.
- It is faster - if there are ~100 atoms of a sample, summing over each pairwise Lennard-Jones interaction, is much slower, than just interpolating the force field from the grid. Because the force evaluation is done several times for each voxel of the 3D scanning grid, it is more efficient to precompute it just once for each voxel, and then interpolate.
- It is more flexible and general - Decoupling of the relaxation process and computation of the force field allows us to plug in any force field. The original motivation was to use an electrostatic force field obtained from a Hartree potential from a DFT calculation. However, it is not limited to that. We can plug in e.g. a derivative of the potential energy of the PP (i.e. Xe or CO ) obtained by scanning it in DFT code, in a similar way as Guo did in JPC C 119, 3, 1483-1488 (2015). The only limitation here is the computational cost of obtaining such a potential from an ab-initio calculation.
If you use ppafm in your research, please cite the latest overview:
And ideally also the original papers: